18017847121

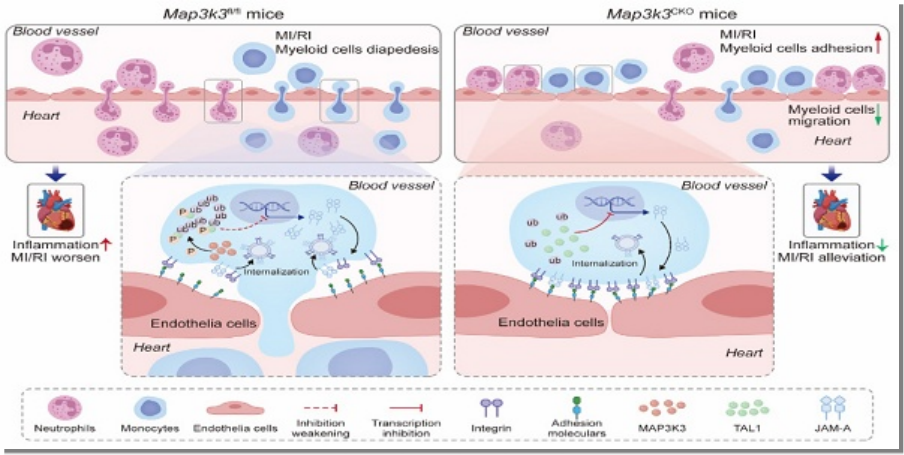
本研究揭示MAP3K3通過TAL1/JAM-A 信號通路調控髓系細胞跨內皮遷移(滲出),在心肌缺血再灌注損傷(MI/RI)中發揮關鍵作用:MI/RI 患者和小鼠模型中,髓系細胞的 MAP3K3 表達顯著上調且與損傷嚴重程度正相關,髓系細胞特異性敲除 Map3K3 可通過增強 TAL1 對 F11r(編碼 JAM-A)的轉錄抑制、減少整合素內化,降低髓系細胞滲出并減輕 MI/RI;Pazopanib(MAP3K3 抑制劑)通過阻斷 MAP3K3 的磷酸化活性,抑制 TAL1 泛素化降解和 JAM-A 表達,減少髓系細胞滲出,顯著改善 MI/RI,為 MI/RI 及相關炎癥疾病提供了新的治療靶點。


研究![]() 臨床背景
臨床背景
急性心肌梗死(MI)后早期血供恢復可減少心肌細胞損失,但繼發性心肌缺血再灌注損傷(MI/RI)仍嚴重威脅心功能;髓系細胞(單核細胞、中性粒細胞)滲出是 MI/RI 炎癥反應的關鍵步驟,通過釋放促炎因子加劇心肌損傷,但該過程的上游調控通路尚未wan全明確。
核心分子
MAP3K3:絲氨酸 / 蘇氨酸蛋白激酶,參與 MAPK、NF-κB 等通路,在炎癥和心血管疾病中起作用,但在髓系細胞滲出中的功能未知。TAL1:轉錄因子,參與造血細胞生成,其在髓系細胞滲出中的作用未明確。JAM-A:由 F11r 編碼,調控整合素內化和白細胞脫黏附,缺失可減少缺血再灌注損傷中的白細胞滲出。
研究目的
闡明 MAP3K3 在 MI/RI 髓系細胞滲出中的作用及分子機制,評估其作為治療靶點的潛力。
研究類型 | 具體內容 | 關鍵參數 / 數量 |
臨床樣本研究 | MI/RI 組:66 例 ACS 患者(PCI 術后 12h,罪犯血管狹窄 > 90%);對照組:6 例冠脈造影陰性者(狹窄 < 50%) | 檢測指標:外周血單核 / 中性粒細胞 MAP3K3 表達、心臟標志物(c Tn特 、CK-MB 等) |
動物模型 | 小鼠 MI/RI 模型:左前降支冠狀動脈結扎 45 分鐘后再灌注 | 實驗動物:6-8 周齡 C57BL/6 景 Map3k3fl/fl、Map3k3CKO 小鼠,性別匹配 |
藥物干預 | Pazopanib(MAP3K3 抑制劑) | 劑量:1.5mg/kg;給藥時間:術后即刻、24h、48h;給藥方式:腹腔注射 |
關鍵實驗技術 | 1. 分子檢測:RNA-seq、磷酸化蛋白質組學、Western blot、雙熒光素酶報告基因 assay;2. 細胞功能:Transwell 滲出實驗、黏附實驗、流式細胞術(髓系細胞表型分析);3. 動物表型:超聲心動圖(LVEF/LVFS 檢測)、Evans blue-TTC 染色(梗死面積)、TUNEL 染色(凋亡) | - |
結果
臨床樣本:MI/RI 患者外周血單核細胞、中性粒細胞中 MAP3K3 的 mRNA 和蛋白水平顯著高于對照組(P<0.05),且與 CK-MB(單核 / 中性粒細胞)、LDH(單核 / 中性粒細胞)、cTn特(單核細胞)等心臟標志物正相關(r 值范圍 0.3-0.5,P<0.05)。
動物模型:小鼠 MI/RI 術后,心臟髓系細胞(CD45+Ly6G + 中性粒細胞、CD45+Ly6G-CD11b+Ly6C + 單核細胞)的 MAP3K3 表達在術后 3 天達峰值,顯著高于對照組(P<0.01)。
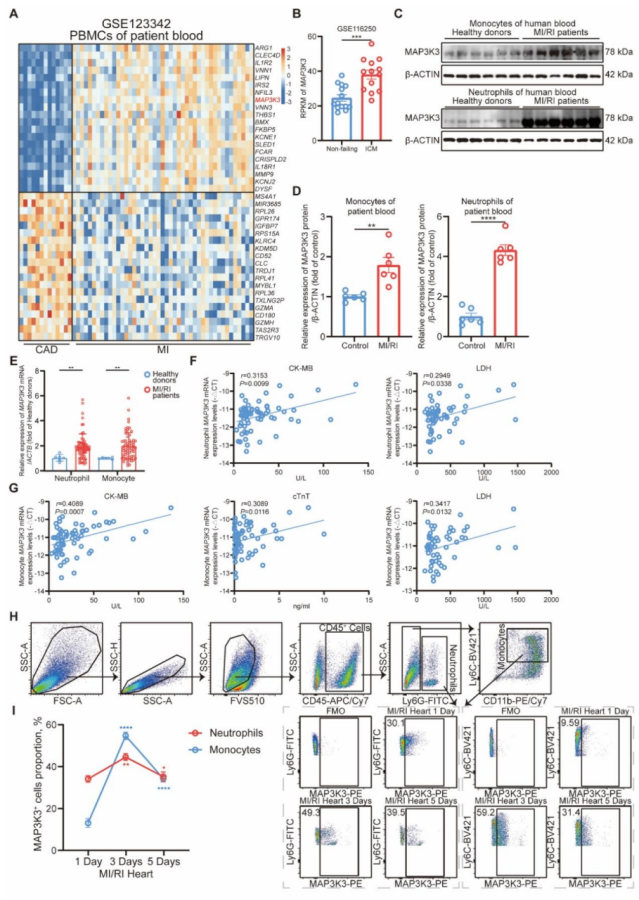
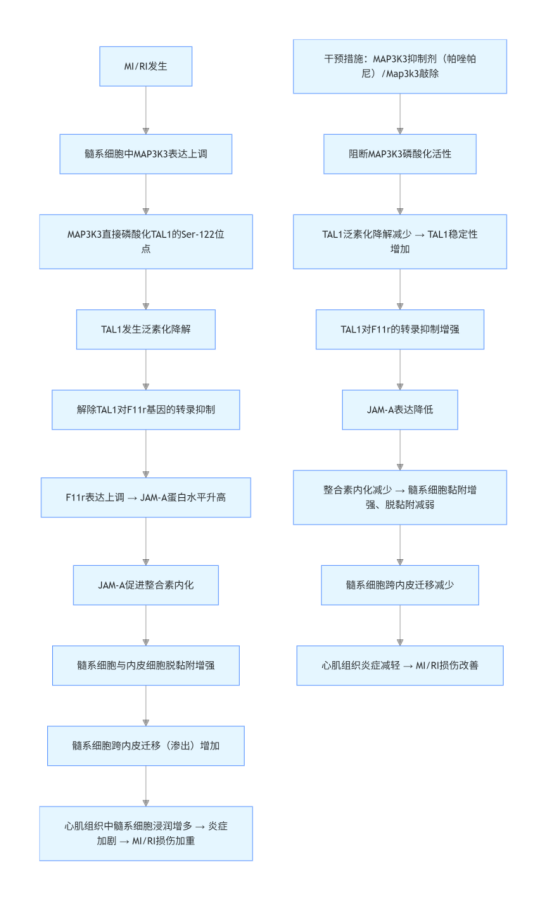
圖 1 心肌缺血再灌注損傷(MI/RI)期間髓系細胞中 MAP3K3 表達上調且與心臟標志物相關。
對 GSE123342 數據集的生物信息學轉錄組分析,發現與穩定型冠心病(CAD)患者相比,MI 患者外周血單個核細胞(PBMCs)中 MAP3K3 表達水平顯著上調。在人類缺血性心肌病(ICM)左心室組織中(GSE57338 數據集),MAP3K3 表達也呈上調趨勢(圖 1B)。在 MI/RI 患者的中性粒細胞和單核細胞中分別驗證了 MAP3K3 的上調表達(圖 1C-E)。MI/RI 患者中性粒細胞和單核細胞中 MAP3K3 表達水平與血清心臟標志物 (包括 CK-MB、cTn特、LDH、pro-BNP、CRP、白細胞介素 - 6(IL-6))及血常規指標的相關性。結果顯示,中性粒細胞和單核細胞中 MAP3K3 的 mRNA 表達水平與 CK-MB、LDH 呈正相關,而單核細胞中 MAP3K3 的 mRNA 表達水平還與 cTn特 呈正相關(圖 1F-G)。
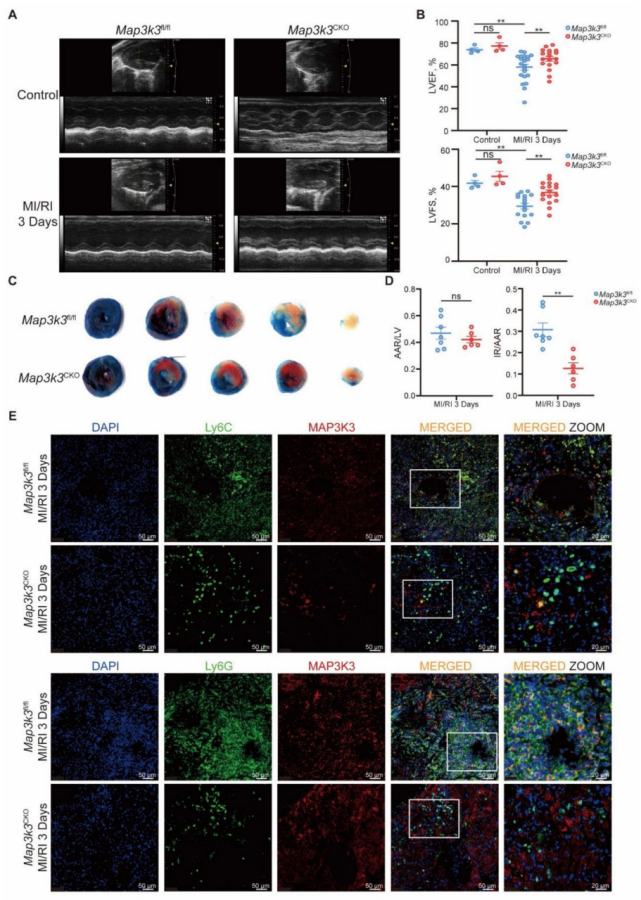
圖 2 髓系細胞特異性 Map3k3 缺陷可減輕心肌缺血再灌注損傷(MI/RI)并減少髓系細胞向心臟的浸潤
構建髓系細胞特異性 Map3k3 缺陷(Map3k3CKO)小鼠,并在 Map3k3CKO 小鼠和 Map3k3fl/fl 小鼠中建立 MI/RI 模型。髓系細胞中 Map3k3 缺陷可改善心臟功能,表現為左心室射血分數(LVEF)和左心室短軸縮短率(LVFS)升高(圖 2A-B),且梗死面積縮小(圖 2C-D)。對 MI/RI 組織切片進行免疫熒光染色,發現髓系細胞中 Map3k3 缺陷可減少心肌組織中髓系細胞的浸潤(圖 2E )。
圖 3 髓系細胞特異性 Map3k3 缺陷可減少心肌缺血再灌注損傷(MI/RI)后髓系細胞從骨髓的跨內皮遷移及體內跨內皮遷移過程
為進一步闡明 Map3k3 缺陷對髓系細胞遷移的影響,我們對 MI/RI 小鼠進行流式細胞術分析。結果顯示,Map3k3CKO 小鼠心臟中浸潤的中性粒細胞和單核細胞比例均降低(圖 3A-B )。追溯髓系細胞的遷移路徑發現,Map3k3CKO 小鼠血液中的中性粒細胞和單核細胞比例降低(圖 3C-D ),而骨髓中的中性粒細胞和單核細胞比例升高(圖 3E-F )。將來自 Map3k3CKO 小鼠或 Map3k3fl/fl 小鼠骨髓的標記髓系細胞混合物注入循環系統,2.5 小時后檢測腹腔和血液中兩種髓系細胞群體的比例(圖 3G)。所有混合物在血液中的標記比例略有差異,證實了 1:1 的混合比例且注射過程無影響。在腹腔中,無論使用何種染料標記,Map3k3CKO 小鼠的髓系細胞比例均降低。而當髓系細胞直接注入腹腔時,兩種小鼠的髓系細胞比例無顯著差異(圖 3H)。
![]()
心功能改善:Map3k3CKO 小鼠 MI/RI 術后 3 天的 LVEF(58.2%±2.1% vs 45.3%±1.8%)和 LVFS(30.1%±1.2% vs 22.5%±1.0%)顯著高于 Map3k3fl/fl 小鼠(P<0.01)。
損傷減輕:梗死面積(IR/AAR)從 Map3k3fl/fl 小鼠的 45.2%±3.1% 降至 Map3k3CKO 小鼠的 28.6%±2.5%(P<0.01),心肌細胞凋亡率(TUNEL 陽性率)降低 35%±4%(P<0.01)。
滲出抑制:Map3k3CKO 小鼠心臟中髓系細胞浸潤比例降低 40%±5%(P<0.01),血液中髓系細胞比例降低 25%±3%(P<0.05),骨髓中髓系細胞比例升高 30%±4%(P<0.01),提示髓系細胞從骨髓到血液的滲出受阻。
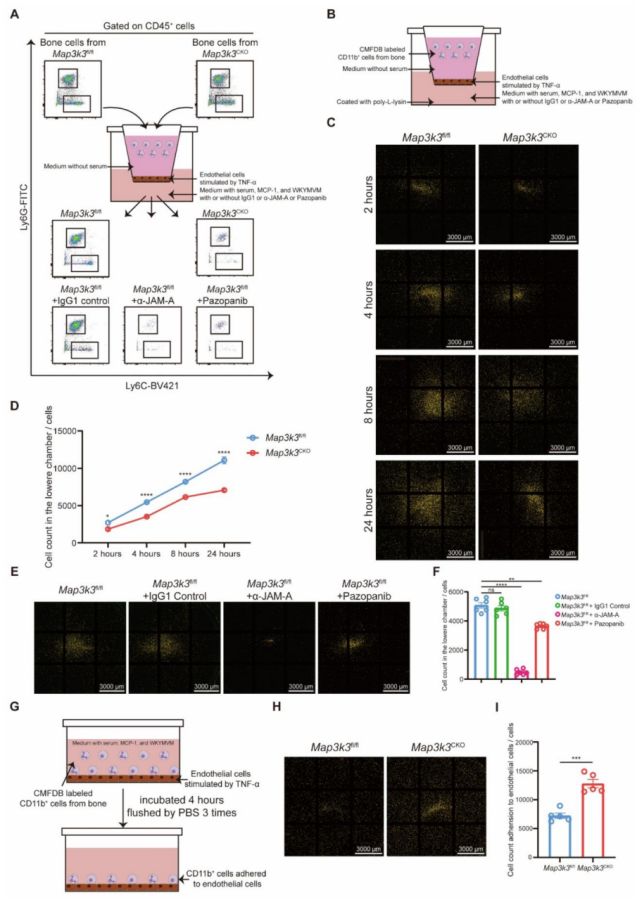
圖4 髓系細胞特異性 Map3k3 缺陷可增強 TAL1 對 F11r 的轉錄抑制功能
測序結果顯示,Map3k3CKO 小鼠中,多種黏附相關基因(如 Itgb3、Adgre4、Itgax)表達上調,而脫黏附基因 F11r 表達下調(圖 4A-B)。黏附與遷移通路的基因本體(GO)分析顯示,Map3k3CKO 小鼠中黏附相關術語的基因富集度升高,遷移相關基因的富集度降低(圖 4C)。KEGG通路 “細胞黏附分子"(mmu04514)的基因分析顯示,Itgb3 和 Itgax 表達上調,F11r 表達下調(圖 4D)。由于 F11r 的表達變化發生在 RNA 水平,我們追溯其轉錄因子,鑒定出四個可能參與 F11r 轉錄調控的候選轉錄因子:TAL1、PRDM1、PPARG 和 PBX1(圖 4E)。在四個候選轉錄因子中,僅 TAL1 在 Map3k3CKO 細胞中的磷酸化水平降低,且特異性發生在 S122 和 S172 位點(圖 4F)。雙熒光素酶報告基因實驗顯示,TAL1 主要抑制 F11r 的轉錄(圖 4G。在 MI/RI 小鼠血液的髓系細胞中觀察到 MAP3K3 和 JAM-A 表達升高,而 TAL1 表達受到抑制(圖 )。在 Map3k3CKO 小鼠中,上述表達模式發生逆轉(圖 4I )。體外實驗中,單核細胞趨化因子 MCP-1 刺激骨髓髓系細胞后,MAP3K3 和 JAM-A 表達升高,TAL1 表達降低;而 Map3k3CKO 小鼠的細胞或經帕唑帕尼(MAP3K3 激酶抑制劑 )處理的細胞中,TAL1 表達升高,JAM-A 表達受到抑制(圖 4J)。在 293T 細胞中(圖 4K),過表達 TAL1 可逆轉 MAP3K3 過表達介導的 JAM-A 上調(圖 4L );而敲低 TAL1 可逆轉 Map3k3 敲除或帕唑帕尼處理介導的 JAM-A 下調(圖 4M-N )。反向實驗證實,MAP3K3 通過下調其抑制性轉錄因子 TAL1,上調 F11r 的表達。由于帕唑帕尼與 Map3k3 敲除具有相似的作用效果,該機制可能依賴于 MAP3K3 的激酶活性。
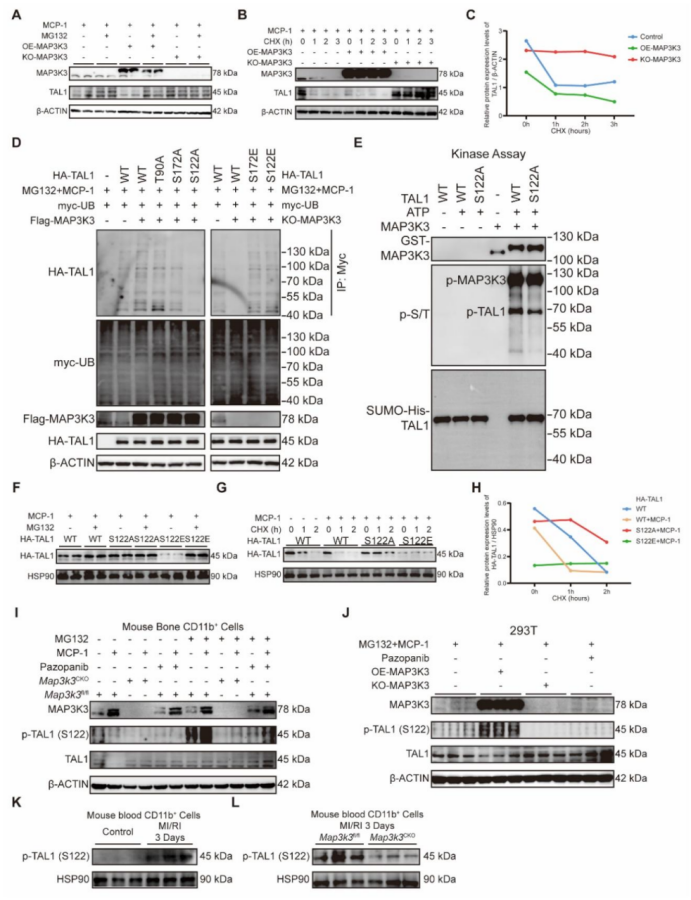
圖 5 Map3k3 缺陷通過 JAM-A 以磷酸化依賴的方式增強髓系細胞黏附并減少跨內皮遷移
與 Map3k3fl/fl 小鼠的髓系細胞相比,Map3k3CKO 小鼠的髓系細胞在整個時間進程中的遷移功能均受損(圖 5A-D)。同時,與未處理或 IgG1 處理的細胞相比,經 JAM-A 抗體或帕唑帕尼處理的細胞遷移能力也降低(圖 5A、E-F)。然而,在黏附實驗中,Map3k3CKO 小鼠的髓系細胞黏附能力增強(圖 5G-I)。
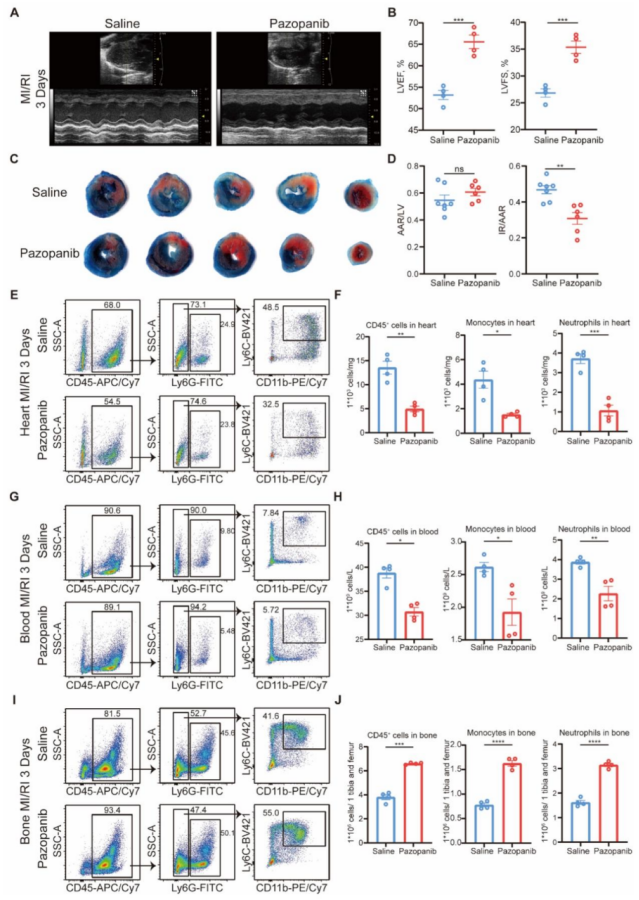
圖 6 MAP3K3 通過在絲氨酸 122(Ser-122)位點磷酸化 TAL1,誘導其蛋白酶體依賴的降解
蛋白酶體抑制劑 MG132 可有效減少 MAP3K3 過表達介導的 TAL1 降解(圖 6A);而使用環己酰亞胺(CHX)抑制蛋白合成后發現,MAP3K3 過表達可加速 TAL1 蛋白降解,Map3k3 敲除可增加 TAL1 蛋白穩定性(圖 6B-C)。這些結果表明,MAP3K3 通過誘導 TAL1 的蛋白酶體依賴降解來減少 TAL1 的表達。MAP3K3 過表達可增加泛磷酸化抗體檢測到的 TAL1 磷酸化水平,并促進 TAL1 的泛素化;T90A 突變對 TAL1 的磷酸化和泛素化無顯著影響,而 S172A 突變和 S122A 突變可顯著降低 TAL1 的磷酸化和泛素化水平。我們進一步將 Ser-122 或 Ser-172 位點突變為谷氨酸(E)以模擬磷酸化狀態,發現 Map3k3 敲除可降低 TAL1 的磷酸化和泛素化水平,而 S122E 或 S172E 突變可維持 TAL1 的泛素化水平(圖 6D)。為明確 MAP3K3 對 TAL1 的直接磷酸化作用,我們進行了體外激酶實驗,發現 MAP3K3 可直接磷酸化 TAL1 的 Ser-122 位點(圖 6E)。S122A 突變可減少 TAL1 的降解,而 S122E 突變可加速 TAL1 的降解,且這種加速降解可被 MG132 逆轉(圖 6F)。使用 CHX 抑制蛋白合成后發現,S122E 突變可加速 TAL1 蛋白降解,而 S122A 突變可增加 TAL1 蛋白穩定性(圖 6G-H)。Map3k3CKO 小鼠的細胞或經帕唑帕尼處理的細胞中,TAL1 的磷酸化水平降低,穩定性增加(圖 6I)。在 293T 細胞系中也觀察到了相同的現象(圖 6J)。在 MI/RI 小鼠血液的髓系細胞中,p-TAL1(S122)的表達水平升高(圖 6K);而在 Map3k3CKO 小鼠中,該表達模式發生逆轉(圖 6L)。
![]()
![]()
心功能與損傷:Pazopanib 處理組小鼠的 LVEF(56.8%±2.0% vs 44.9%±1.9%)、LVFS(29.5%±1.1% vs 22.3%±1.1%)顯著高于生理鹽水組(P<0.01),梗死面積(IR/AAR)從 44.8%±3.0% 降至 30.2%±2.6%(P<0.01)。
滲出抑制:Pazopanib 處理后,心臟、血液中髓系細胞比例分別降低 38%±4%、23%±3%(P<0.01),骨髓中髓系細胞比例升高 28%±3%(P<0.01),與 Map3k3CKO 小鼠表型一致。
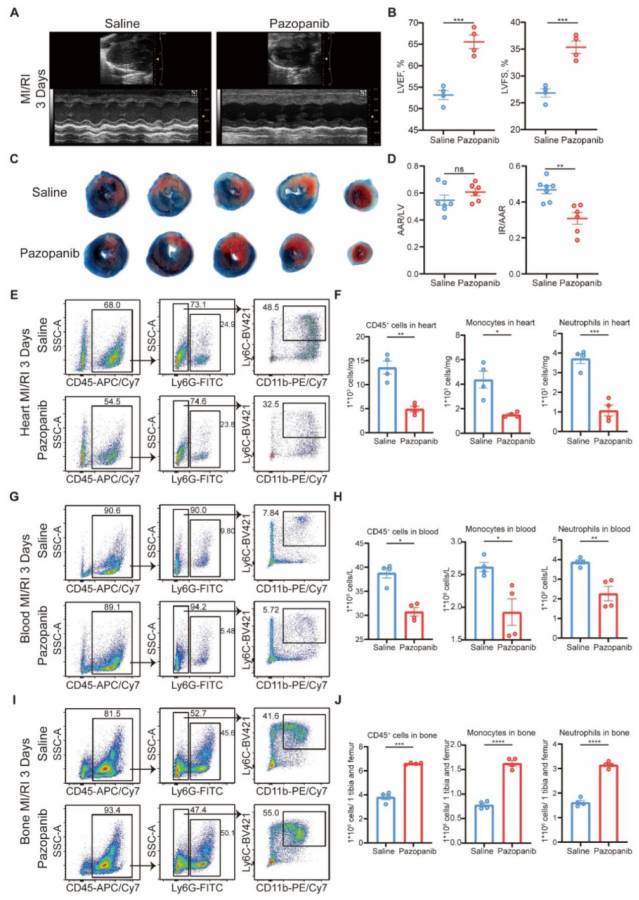
圖 7 帕唑帕尼通過降低 MAP3K3 的磷酸化活性及髓系細胞從骨髓的跨內皮遷移,改善心肌缺血再灌注損傷(MI/RI)
帕唑帕尼治療可改善 MI/RI,表現為 LVEF 和 LVFS 升高(圖 7A-B)、梗死面積縮小(圖 7C-D)以及 TUNEL 染色顯示的心肌細胞凋亡減少。對 MI/RI 小鼠的流式細胞術分析顯示,帕唑帕尼治療可減少中性粒細胞和單核細胞從骨髓向心臟(圖 7E-F)和血液(圖 7G-H)的遷移(圖 7I-J)。
研究結論與意義
核心結論:MAP3K3 通過磷酸化 TAL1(Ser-122)促進其泛素化降解,解除對 F11r 的轉錄抑制,上調 JAM-A 表達,增強整合素內化,最終促進髓系細胞脫黏附和滲出,加劇 MI/RI。
臨床意義:Pazopanib(低劑量、短期給藥)可通過抑制 MAP3K3 活性,阻斷上述通路,減少髓系細胞滲出,為 MI/RI 及其他炎癥相關疾病(如膿毒癥)提供了新的治療策略。
 歡迎來到
歡迎來到 聯系人:張
聯系人:張 地址:上海市長江南路180號長江軟件園B區B637室
地址:上海市長江南路180號長江軟件園B區B637室 郵箱:2844970554@qq.com
郵箱:2844970554@qq.com 傳真:
傳真: